










Original Articles

DOI: 10.24953/turkjpediatr.2025.5613
Pages: 135-143
DOI: 10.24953/turkjpediatr.2025.5653
Pages: 144-152
DOI: 10.24953/turkjpediatr.2025.5528
Pages: 153-161
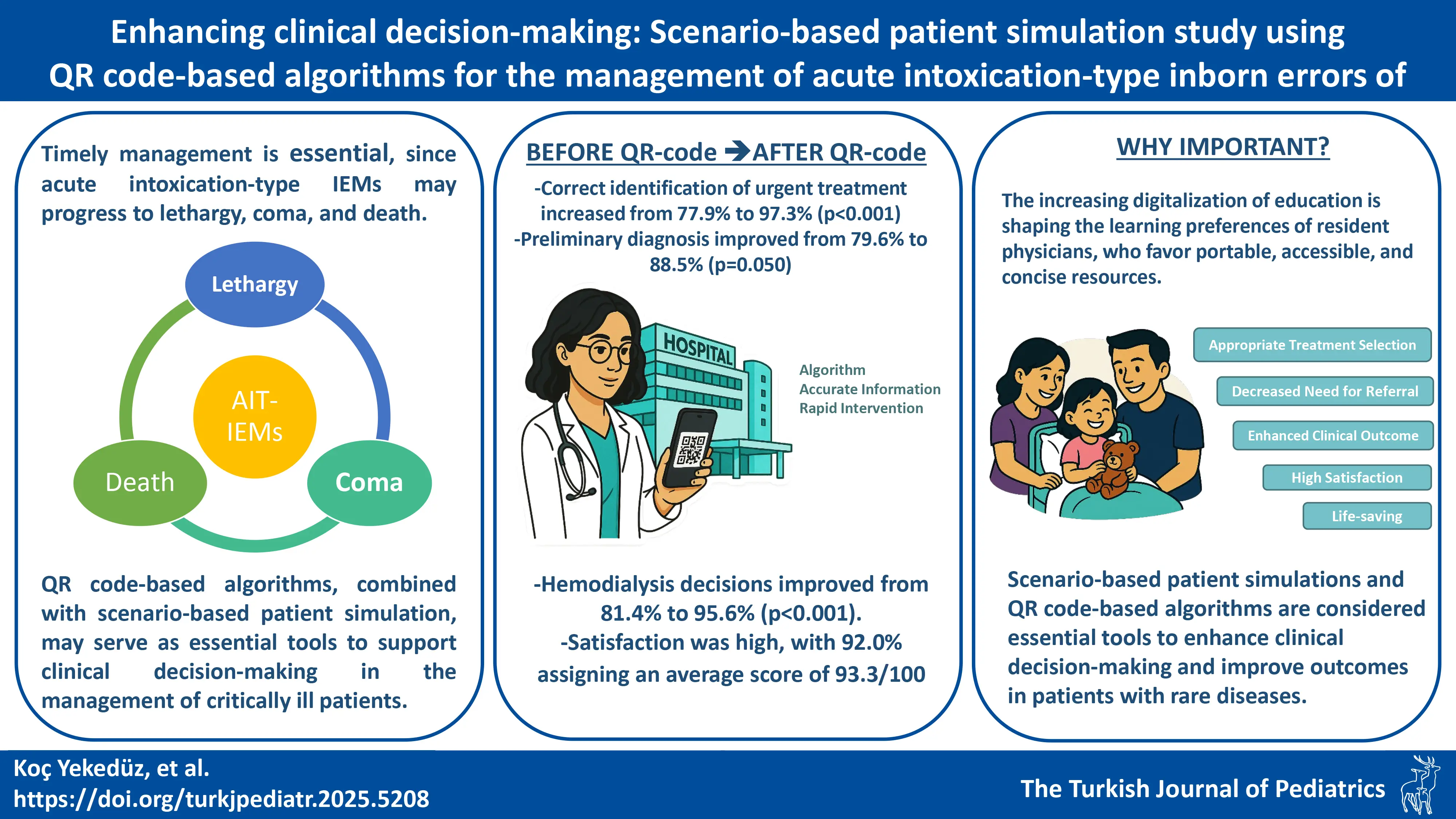
DOI: 10.24953/turkjpediatr.2025.5208
Pages: 162-174

DOI: 10.24953/turkjpediatr.2025.5263
Pages: 175-185
DOI: 10.24953/turkjpediatr.2025.4805
Pages: 186-194
DOI: 10.24953/turkjpediatr.2025.5293
Pages: 195-207
DOI: 10.24953/turkjpediatr.2025.5460
Pages: 208-220
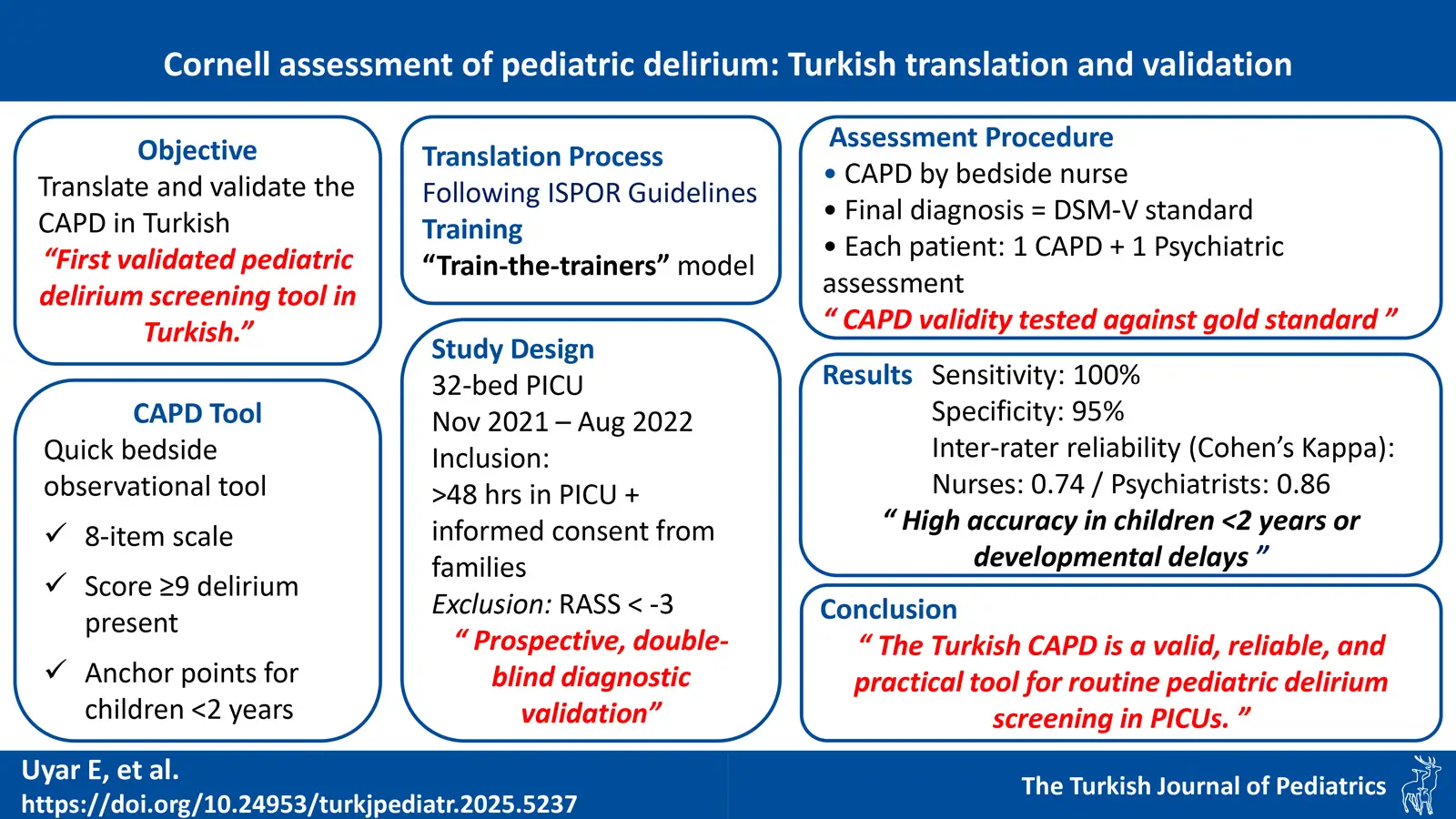
DOI: 10.24953/turkjpediatr.2025.5237
Pages: 221-229
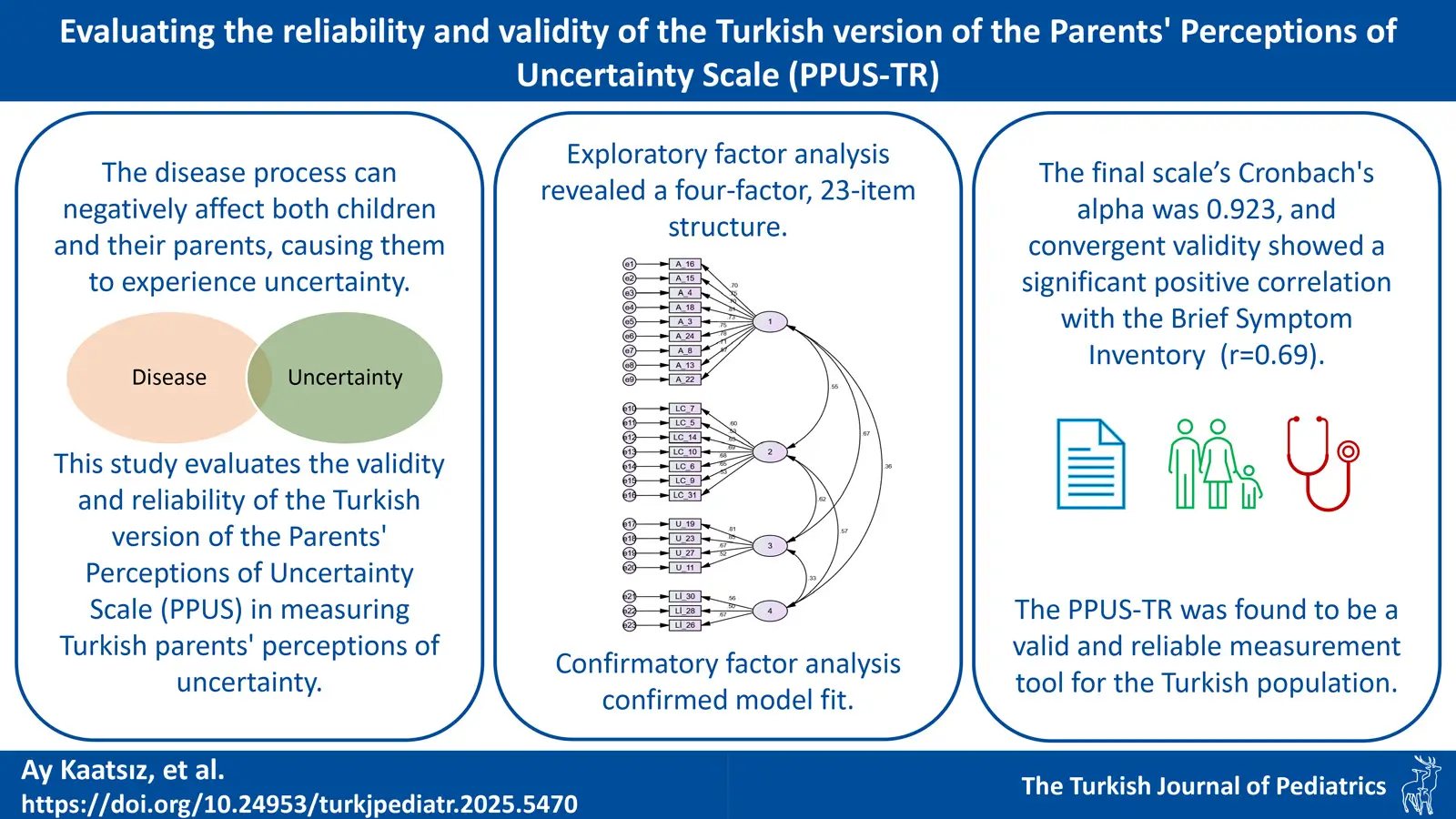
DOI: 10.24953/turkjpediatr.2025.5470
Pages: 230-241
Short Communications
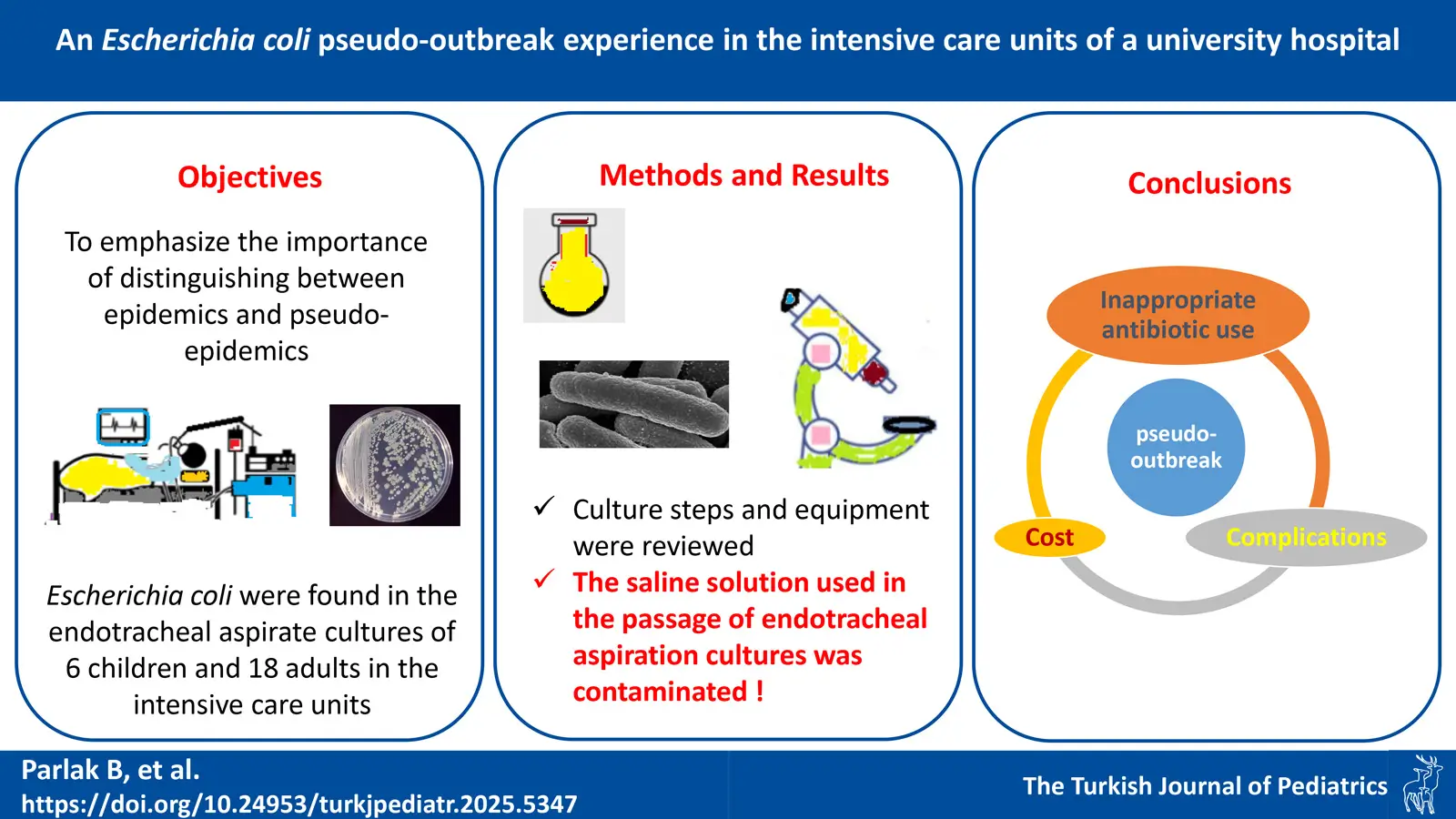
DOI: 10.24953/turkjpediatr.2025.5347
Pages: 242-247
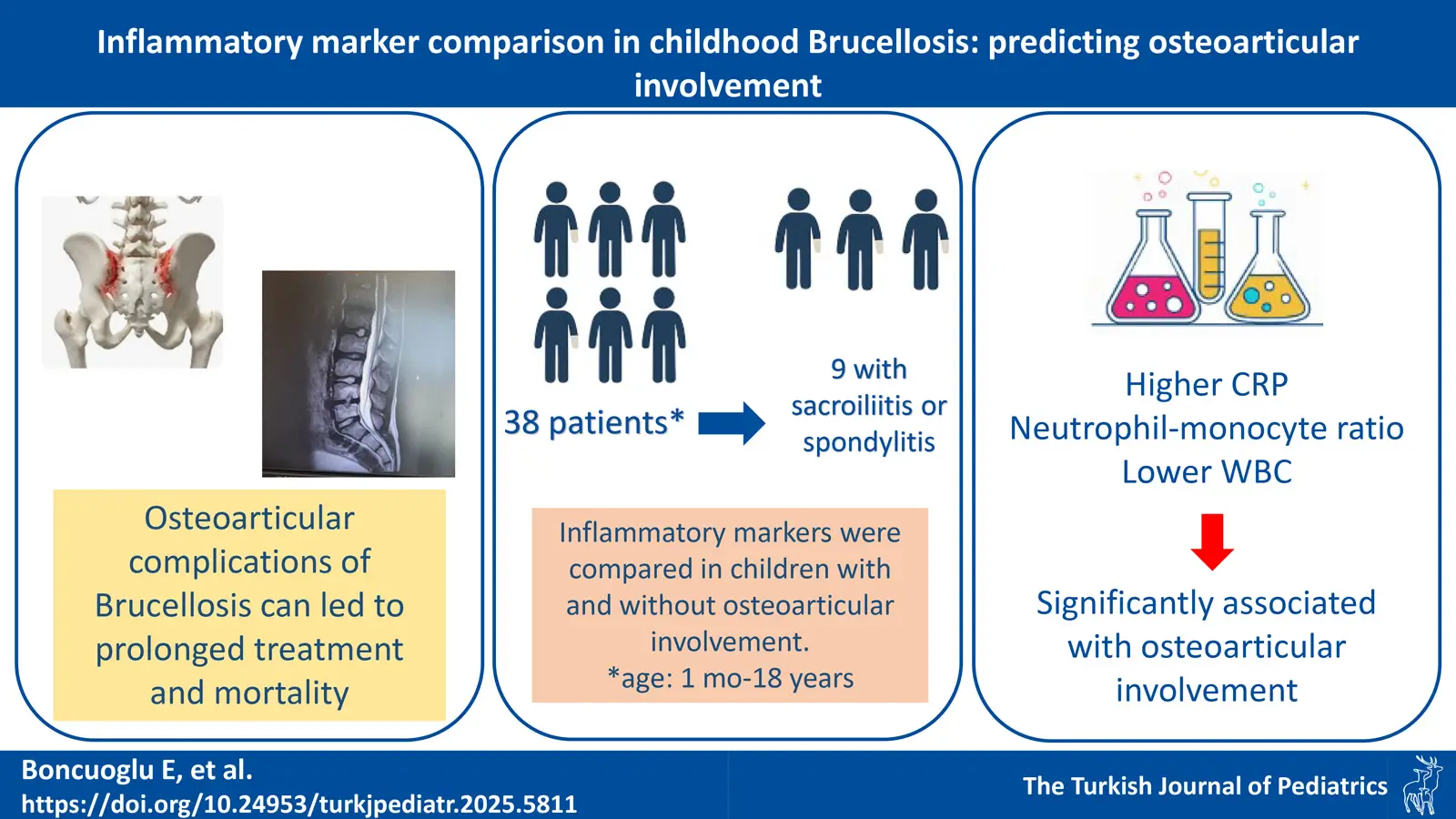
DOI: 10.24953/turkjpediatr.2025.5811
Pages: 248-253
Case Reports
DOI: 10.24953/turkjpediatr.2025.5329
Pages: 254-258
DOI: 10.24953/turkjpediatr.2025.5786
Pages: 259-267
DOI: 10.24953/turkjpediatr.2025.5488
Pages: 268-272
DOI: 10.24953/turkjpediatr.2025.5382
Pages: 273-281
DOI: 10.24953/turkjpediatr.2025.5133
Pages: 282-287
Letters to the Editor
DOI: 10.24953/turkjpediatr.2025.5578
Pages: 288-289